7 min read
It’s not often that small biotechnology companies pursue basic research into disease mechanisms. However, the serious impact of the SARS-CoV-2 outbreak on our families, friends, and community has prompted us to seek solutions for solving the COVID-19 pandemic. At Antibody Solutions (AS) and Single Cell Technology (SCT), we sought answers to the nature of the virus infection using our tools at hand.
In results published at Preprints 2020, 2020050407 (doi: 10.20944/ preprints202005.0407.v1), we present what our team uncovered through a viral genome sequencing and molecular modeling study. We believe it sheds new light on potential reasons why a significant mutant of SARS-CoV-2 — D614G, the mutation believed to have first appeared in Europe and subsequently migrated to the East Coast of the United States — shows a competitive advantage over the reference genotype of SARS-CoV-2 cases in China.

Chun-Nan Chen of SCT, and his colleagues Joy Tang and Allison Schulkins, were inspired to mine worldwide SARS-CoV-2 genome data with the company’s informatics platform to identify SARS-CoV-2 mutants, their geographical distribution and appearance over time. With my colleague, Kurt Deshayes, we used our suite of homology modeling tools to understand how mutations could impact the structure and function of the SARS-CoV-2 spike protein. With so much unknown about the transmission and virulence of the virus, we hoped our efforts would aid in the basic understanding of its biology.
The rise and dominance of the D614G mutation.
Chun-Nan and his team analyzed 11,542 viral genome records from the GISAID database collected through April 27, 2020. (This is the largest cohort of sequence samples that I’ve seen analyzed to date.) They identified mutations by comparing each sample to a sequence originally reported from Wuhan China. They found mutations in the Spike or “S” protein at 103 positions. D614G was, put simply, the dominant mutation, occurring in 56% of all sequences — far more than all of the other identified mutations combined. Looking at the geographic distribution, the D614G mutation predominated in European countries and most of the U.S. states. A few U.S. states, principally Washington and California, had more samples with the genotype of the original Wuhan virus (see select data in Figure 1).
Focusing on the D614G mutation, their research included a comparative analysis of daily cumulative genotype counts by country and by U.S. states, This allowed them to examine which protein sequence outpaced the other as samples were collected in each geography over time. They found a dramatic increase in the frequency of the G614 mutant over time in multiple regions.
What’s really striking is that when both arrived at the same time, the mutant surpassed the Wuhan virus genotype in many states, suggesting the virus having the G614 spike protein virus outcompetes the Wuhan D614 virus. The Wuhan virus genotype dominated when it had a “head start”; in Washington and California, for example, dominance of the Wuhan virus was due to much earlier arrival compared to the D614G mutant.
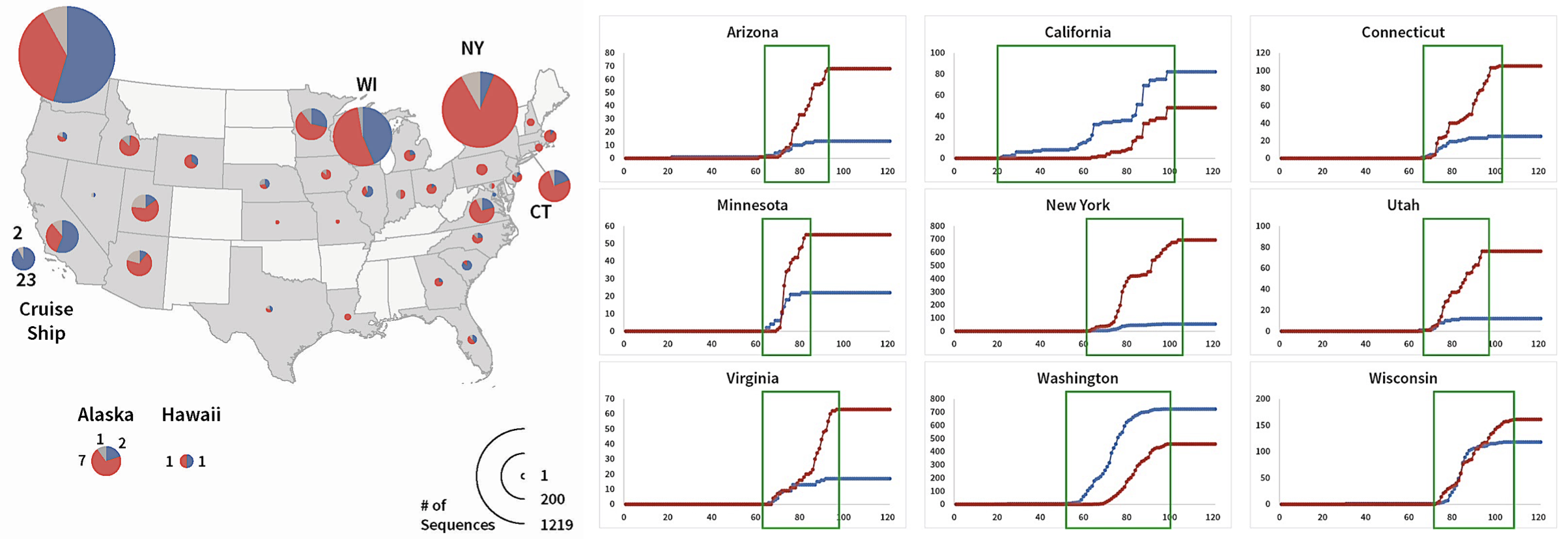
FIGURE 1. S Protein genotype totals and daily cumulative genotype counts.
Frequencies of S protein mutations by state are shown on left and cumulative counts are shown on right. Red is D614G, Blue is Wuhan, and Gray are other mutations (shown in left figure only). In the right figure the x-axis is the day of sampling, and the Y axis is the cumulative total for the genotype. Day 0 is January 1, 2020. From Preprints 2020, 2020050407 (doi: 10.20944/preprints202005.0407.v1).
Molecular modeling reveals unexpected changes in D614G structure in the furin binding domain.
My colleague Kurt Deshayes and I at Antibody Solutions had been busy homology modeling domains of the S protein for new antibody discovery programs. The polypeptide domain surrounding the D614G mutant had yet to be identified as having a function in virus infectivity and didn’t seem to be on anyone’s “radar screen.” So when Chun-Nan contacted me regarding his observations, we thought it would be interesting to look at regions adjoining the D614G mutation. We were curious to determine if a single amino acid substitution would impart a significant change in spike protein structure.
The closest functional domain of the S protein to the D614G mutation is a furin cleavage site. Furin cleavage of the S protein is an important mechanism leading to virus infection. However, the D614G mutation lies approximately 68 amino acids and 24 angstroms away from the furin cleavage domain.
We had our doubts that a single amino acid substitution would have an effect at a long distance, but it had been reported in the literature to occur for other proteins. When we looked at existing Cryo-EM (electron microscopy) structures of SARS-CoV and SARS-CoV-2, we got “blanks”; Cryo-EM didn’t resolve the structure of the site and, in turn, couldn’t shed any light on the secondary structure at the site.
It is computationally challenging to model very large proteins like the full S protein, which is 1,261 amino acids long in its mature form. We chose to model a region of the protein from amino acids S591 to N710 containing the D614G mutation and the furin cleavage domain of SARS-CoV-2 (R682-S686). Luckily, the secondary structure in the vicinity of the two sites had been largely defined through Cryo-EM structures and provided a valid starting point for homology modeling.
A multiple template threading mechanism, I-TASSER, was used to create models of the S591 to N710 region. We created two models with the only difference being a change in amino acids at position 614 (D614 and G614, respectively). We observed a provocative result in which the only significant change in structure was at the furin cleavage domain: The G14 model showed the furin domain as an alpha helix, and the D614 as random coil. The secondary structure algorithm, KSDSSP (the Kabsch and Sander algorithm for defining secondary structure of proteins), was also found to predict the same differences in secondary structure. (See Figure 2 below)
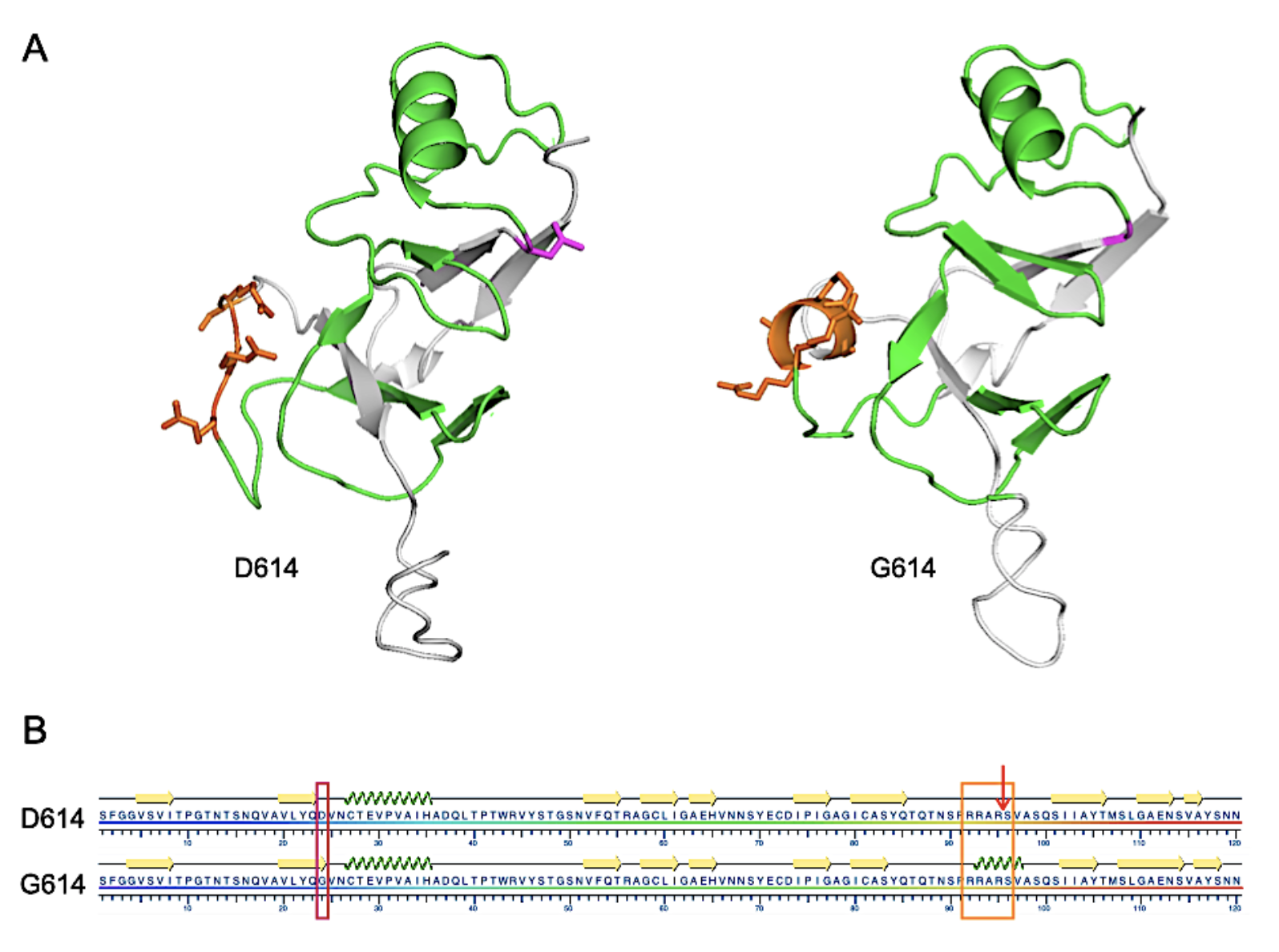
FIGURE 2. Structural representations of D614 and G614 SARS-CoV-2 S protein from S591 through N710.
A. I-TASSER-derived models. The furin cleavage domain 682-RRARS-686 is shown in orange and D614 (left) or G614 (right) are shown in magenta. Residues between 614 and the cleavage domain are shown in green.
B. Results of the KSDSSP secondary structure algorithm.The D614G mutation is highlighted with a magenta box, the furin cleavage domain with an orange box and the cleavage site by the arrow. Beta sheet or alpha helix secondary structures are indicated by yellow arrows or green corkscrew, respectively. From Preprints 2020, 2020050407 (doi: 10.20944/preprints202005.0407.v1).
Next, we were curious if the change in the furin domain secondary structure between the D614 and G614 models might translate into a more favorable orientation for cleavage by furin.
Furin cleaves between R685 and S686 of the cleavage domain 682-RRARS-686. A rate-limiting step in cleavage by furin is the placement of R685 and R682, the P1 and P4 enzyme substrate residues, respectively, into the corresponding binding pockets. Since no one has been able to obtain a crystal or Cryo-EM structure of a protein substrate in the furin catalytic site, we used the known structure of a small-molecule inhibitor bound to furin as a template. We found the D614G mutation, through the conformational change, better orients the P1 and P4 residues with the orientation of the inhibitor (see article) and within the active site of furin (Figure 3).
Thus our modeling results predict the D614G mutant spike protein would act as a better substrate for furin cleavage. With that in mind, if furin cleavage is more efficient for the D614G mutant, this would give it an advantage over the Wuhan virus for infectivity.
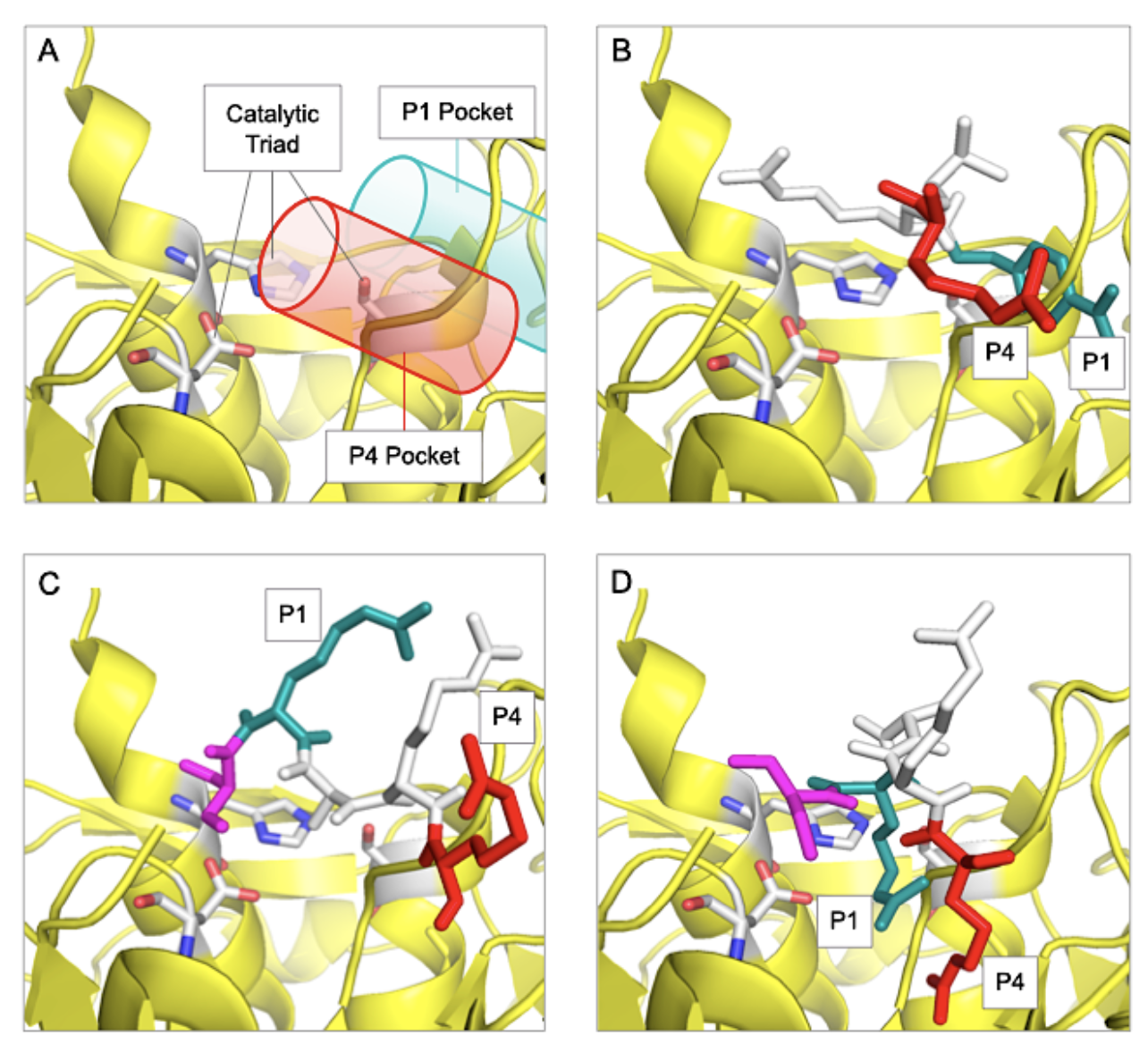
FIGURE 3. Alignment of D614 and G614 in the catalytic domain of furin.
A. The catalytic domain of furin is shown in yellow (panels A-D) with the locations of the P1 (green) and P4 (red) binding pockets shown as cylinders. B. Alignments of Inhibitor, Meta-guanidinomethyl-phenylacetyl-RVR. C. D614 682-RRARS-686. D. G614 682-RRARS-686.
Implications and next steps
Our proposed mechanism for how the D614G mutation could lead to increased infectivity certainly does not preclude other mechanisms that could act in concert with the one we’ve proposed here.
It’s quite possible, for example, that the D614G mutation could influence other critical processes so that the overall change in rate of infection is greater than is possible with a modification of the rate of a single event. It’s still crucial to measure the relative rates of furin cleavage of the D614G versus Wuhan S protein and how that impacts the infection process. There has also been recent speculation in the popular press regarding the role of mutations on morbidity and mortality of Covid-19. What impact the D614G mutation would have on these aspects of COVID-19 remain to be determined.
This is a case where sequencing and molecular modeling working hand-in-hand has helped build a greater understanding of the pathogenesis of SARS-CoV-2. Most mutations either fade from view or, if they stick, don’t have deleterious effects.
But D614G is different. This mutation appears to have a competitive advantage over the Wuhan virus in its spread; and equally important, there’s a plausible mechanism to explain how it seized that advantage. Our results suggest the furin cleavage domain could be a key target for therapeutic antibodies, vaccines and other modalities.
We hope our observations inspire other researchers to further investigate the impact of the D614G mutation on SARS-CoV-2 pathogenesis, and we welcome any opportunity to collaborate on this work. In addition to this study, our team is advancing a range of other SARS-CoV-2 research initiatives, both on our own and with clients, to pursue vaccines and antibody therapies. Right now, it’s hard to imagine a more impactful and far-reaching endeavor to benefit our families, our communities and our world.
If you want to learn more about this study or how our molecular modeling expertise and other capabilities can help move your research forward more quickly, please don’t hesitate to contact me here.







