9 min read
Antibody Solutions has been tracing the evolution of SARS-CoV-2 from the earliest days of the pandemic. As part of our program developing antibodies to prevent spike protein cell entry via cleavage at the S1/S2 site by furin, we’ve followed the evolution of the S1/S2 sequence as it emerged from one dominant variant to the next. We’ve observed that the Delta variant was likely the most infectious variant of SARS-CoV-2 to date, and we offered our predictions that subsequent Omicron variants – dominant for most of 2023 – would be “highly transmissible, but with lower disease severity,” a prediction that turned out to be correct, both in broad reports and in studies of small patient groups.
We’re now faced with the rise of the tenacious new dominant SARS-CoV-2 variant JN.1. This strain – which was first detected in September of last year – accounted for 86% of U.S. cases as of January 20, 2024, and was believed to be present in as many as 96% of cases in the U.S. by mid-February. Simply put, if you come down with COVID-19 in the United States these days, it’ll almost certainly be the JN.1 variant. The World Health Organization (WHO) reports that JN.1 has been found in as many as 71 countries, accounting for 66% of global cases. Taking into account the information available about this current strain of SARS-CoV-2, and juxtaposing that intel against the virus’ history of previous strains, we feel that we’re well-situated to provide analysis of how JN.1 differs from previous variants.
Old Positions and New Mutations
SARS-Cov-2’s notable variants all share the common S1/S2 site sequence RRARS, with cleavage by furin occurring between the arginine (R) and serine (S) residues. Where the variants differ are the mutations exhibited just upstream of RRARS. These mutations confer different conformations on the RRARS domain, making the S1/S2 site either more or less favorable for cleavage by furin. This mechanism of action has been predicted by our molecular modeling and confirmed by outside observations. The JN.1 variant incorporates a mutation at the S1/S2 site, H681R. This specific mutation was first seen in the Delta variant, indicating that it confers a more favorable conformation for furin cleavage, as compared to the Omicron variant.
Mutations at the S1/S2 site as well as other mutations give the variant a competitive advantage. As shown in Figure 1, Delta, Omicron, and JN.1 all have a mutation at position 681 that changes the original proline (P) to either arginine (R) in Delta and JN.1, or histidine (H) in Omicron. A second mutation of asparagine to lysine (N679K) is seen in both Omicron and JN.1.
Figure 1: Sequences and Periods of Dominance for Delta, Omicron and JN.1 Variants
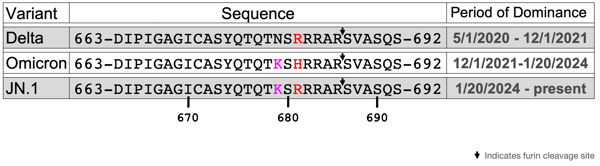
Table depicting the amino acid sequences proximal to the S1/S2 cleavage site for Delta, Omicron, and JN.1, accompanied by the dates that the variant was most prevalent. The furin cleavage site is indicated by a downward arrow and mutations from the parent virus are in pink and red.
Late last year, the virus’ evolution changed course from HV.1 to BA.2.86, and finally to JN.1. Looking at the phylogenetic tree of mutations (Figure 2A), one can see that the Omicron variants XBB.1.5 and HV.1 reside on distinctly different branches than Delta. A result of this evolutionary detour is the return of the H681R mutation first observed in Delta (Figure 2B). One reasonable hypothesis for H681R’s reemergence: this mutation, occurring independently in two different evolutionary pathways, confers some sort of survival advantage on the virus.
Figure 2A: Phylogenetic Tree of Delta, Omicron and JN Variants
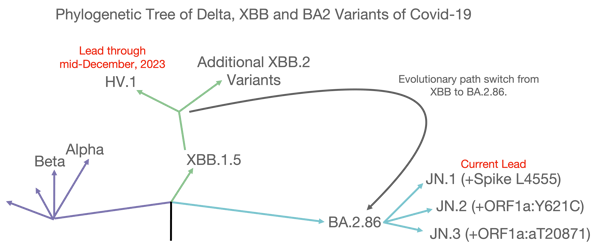
Phylogenetic tree graph showing the evolution of SARS-CoV-2 variants. The Delta path is shown in purple, the XBB.1.5 path in green, and the most recent BA.2 path in blue. This figure has been adapted with permission from a figure generated by Eric Topol and has been simplified to illustrate the evolution of the most relevant variants to this discussion.
Figure 2B: SARS-CoV-2 Spike Sequence and Mutation Map with Leads
%20copy-003%20(1).png?width=600&height=334&name=JN-1%20blog%20figures%201%2c2A%2c2B%2c3A%2c3B%2c3C%20(updated%202-29-24)%20copy-003%20(1).png)
Spike sequence and mutation map with tiles representing mutations as they occur in each variant. Purple tiles indicate the presence of the mutation, while white tiles represent the absence of the mutation. This has been adapted with permission from a figure generated by Eric Topol and has been simplified to illustrate the evolution of the most relevant variants to this discussion.
To obtain insight into the return of H681R found in JN.1, we modeled the docking of Delta, Omicron, and JN.1 S1/S2 site variants to the furin active site using the ColabFold version of AlphaFold Multimer. In each case, 29-mer segments of the S1/S2 region were used to generate large binding interfaces that are more amenable to modeling in AlphaFold Multimer. The models were generated with no alignment bias of the substrate in relation to the enzyme. The docking models for Delta and JN.1 place the arginine in the P1 position, adjacent to the active site. This positions S and R such that the connecting bond is cleaved by the enzyme, which in turn aligns with the established mechanism of furin cleavage.
Figures 3A and 3B: Furin S1/S2 Configurations in Delta and JN.1
-004.png?width=600&height=302&name=JN-1%20blog%20figures%201%2c2A%2c2B%2c3A%2c3B%2c3C%20(updated%202-29-24)-004.png)
Figure 3A (Delta)
-005.png?width=600&height=301&name=JN-1%20blog%20figures%201%2c2A%2c2B%2c3A%2c3B%2c3C%20(updated%202-29-24)-005.png)
Figure 3B (JN.1)
In these figures, the S1/S2 regions Delta (3A) and JN.1 (3B) are docked to furin, with furin shown as a space-filling model. The carbons are shown as white, nitrogen as blue, and oxygen as red. The S1/S2 region is shown as a stick model with different colors representing carbon (green), nitrogen (blue), and oxygen (red). Key amino acids and their positions in the sequence are indicated.
The situation changes for SARS-Cov-2’s Omicron HV.1 variant. This time, the most stable model leaves the P1 binding pocket empty, shifting the residues away from the active site by two residues taking the reactive R and S outside the active site. This suggests that, when compared to Delta or JN.1, HV.1 is less optimized for furin processing.
Figure 3C: Furin S1/S2 Configurations in Omicron
-006.png?width=600&height=301&name=JN-1%20blog%20figures%201%2c2A%2c2B%2c3A%2c3B%2c3C%20(updated%202-29-24)-006.png)
Figure 3C HV.1
In this figure, the S1/S2 region of Omicron is docked to furin, with furin shown as a space-filling model. The carbons are shown as white, nitrogen as blue, and oxygen as red. The S1/S2 region is shown as a stick model with different colors representing carbon (green), nitrogen (blue), and oxygen (red).
The evolutionary path switch to JN.1 corrects the poorer fit of the Omicron variants S1/S2 site into the furin catalytic site. This would be expected to enhance viral entry into the cell. While the virtual models shown in Figures 3A-C don’t provide atomic resolution of the S1/S2 furin interaction, they do provide a rationale as to why the virus course-corrected to evolve JN.1. This would also, at least in part, explain why JN.1 has so rapidly become the dominant variant.
It is too early to determine if the differences between JN.1 and Omicron leads to more severe COVID-19 infection. Early indications from the Center for Disease Control (CDC) suggest that it does not generate a more severe response. The meteoric rise in prevalence of this strain, however, leaves no question that it is more transmissible than Omicron. Regardless of the severity of the illness it causes, the JN.1 mutation absolutely poses an increased risk to the population.
Where Do We Go From Here?
It’s important to track SARS-CoV-2 as it evolves and to examine how its mutations impact the course of the pandemic. There is compelling evidence that blocking S1/S2 cleavage by furin can prevent the development of severe disease, thereby reducing the likelihood of transmission. For this blocking to occur, an antibody must potently bind the S1/S2 site of the SARS-CoV-2 variant responsible for the infection.
While tracking the evolution of SARS-CoV-2, Antibody Solutions created humanized antibodies that bind to the S1/S2 site of the spike protein in order to block furin processing and viral entry into the cell. We were able to accomplish this using our Cellestive discovery platform. Cellestive encompasses an array of flexible discovery routes and is run by a highly skilled team. It’s this blend of expertise and infrastructure that led us to discover the importance of the S1/S2 site. This same comprehensive and collaborative approach can be harnessed to tackle any of your discovery projects (the more challenging, the better!). Reach out to us to get our team exploring and experimenting on your behalf.





